
Alain Delacroix
Professeur honoraire, chaire « Chimie industrielle – Génie des procédés » du Conservatoire national des arts et métiers
L’isolement de la quinine par Pelletier et Caventou en 1820 est un grand progrès dans la lutte contre le paludisme car il permet de délivrer une dose de molécule active reproductive et d’en déduire une posologie efficace. Avant, la dose administrée au patient dépendait du type de l’écorce de quinquina et l’effet allait de l’inefficace au toxique.
Compte tenu de l’importance de la maladie, de nombreuses recherches sont alors initiées sur la molécule elle-même mais aussi sur les substituts possibles. Cela est d’autant plus nécessaire que la quinine, comme tout médicament actif, est toxique et n’est pas efficace sur tous les plasmodiums.
En 1849, Adolf Strecker donne la formule brute de la quinine : C20H24N2O2 et en 1853, Pasteur, en travaillant sur l’acide tartrique et en utilisant des amines chirales, trouve une molécule proche de la quinine, la quinotoxine.
A la fin du XIXe siècle, Zdenko Skraup donne la structure chimique de la quinine et l’on constate qu’elle est complexe car elle comporte au moins un carbone asymétrique, d’où plusieurs isomères. Ceux-ci n’ont forcément pas la même activité, ce qui implique une synthèse stéréosélective.
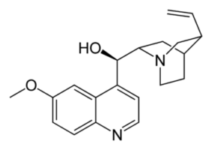
Au début du XXe siècle, Paul Rabe et Carl Kindler publient la synthèse de la quinine à partir de la quinotoxine. Le 11 avril 1944, Robert Woodward et William von Eggers Doering, de Harvard, décrivent la synthèse de la quinine, ce qui fait grand bruit à l’époque, mais elle est sujette à controverse. La synthèse totale et stéréosélective est effectuée par Gilbert Stork en 2001. La vingtaine d’étapes nécessaires, toutefois, implique un rendement très faible et ne permet pas l’industrialisation du procédé.
L’usage de la quinine décline jusqu’aux années soixante-dix puis reprend en raison de la résistance de souches de plasmodium aux nouvelles molécules.
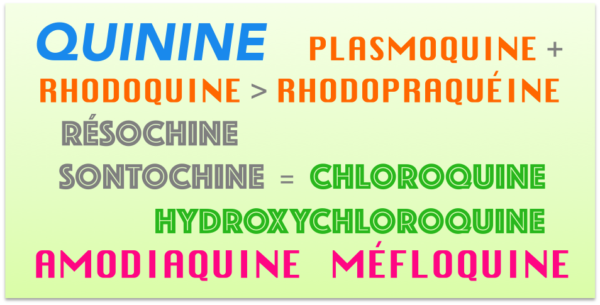
Les recherches sur les antipaludéens de synthèse commencent à la fin du XIXe siècle. A cette époque, on constate qu’un colorant, le bleu de méthylène, a une action contre la syphilis et certains microbes. En 1891, Paul Ehrlich remarque l’action antimalaria de ce colorant mais son effet, justement très colorant, est gênant pour les malades. Une équipe de Bayer synthétise et teste alors des quantités de molécules de structure voisine et remarque, en 1925, qu’une aminoquinoléine est efficace et la dépose sous le nom de plasmoquine. La formule reste secrète jusqu’en 1928.
Pendant ce temps, les chercheurs français et britanniques subodorent que certains dérivés de la quinoléine ont une action antipaludique. En 1930, Ernest Fourneau et son équipe de l’Institut Pasteur synthétisent la rhodoquine, une aminoquinoléine, qui est plus efficace que la plasmoquine. La rhodoquine, associée à la plasmoquine sera très utilisée en France jusqu’aux années quatre-vingt sous le nom de rhodopraequine.
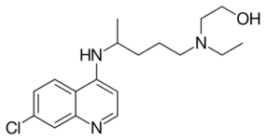
En 1932, on produit l’atabrine, qui est une 9-aminoacridine, molécule efficace mais qui a le défaut de jaunir la peau des patients. Hans Andersag d’IG Farben remarque, en 1934, que le diphosphate de chloroquine, appelé résochine, est un bon antipaludéen mais est trop toxique. Il synthétise alors diverses molécules de structures comparables et observe que la 3-méthylchloroquine, appelée sontochine, est efficace et moins toxique. Juste avant la deuxième guerre mondiale, les Allemands brevètent la résochine et la sontochine et, en 1941, Bayer accorde à Winthrop les droits sur ces deux produits. Le fait que les différentes armées sont alors décimées par la malaria accélère la recherche et de nombreux travaux sont effectués sur les aminoquinoléines. En 1941, Rhône Poulenc Spécia passe un accord avec IG Farben pour faire des essais sur la sontochine et, en 1942, le docteur Philippe Jean Decourt effectue des tests à Tunis, alors sous domination allemande. Le 7 mai 1943, la première armée britannique entre à Tunis et le docteur Schneider, qui connaît les résultats de Decourt, les communique aux alliés.
En février 1946, la sontochine prend le nom de chloroquine et devient un célèbre médicament antipaludéen en 1947. En France, elle prend le nom de Nivaquine en 1949. L’industrie pharmaceutique hésite cependant à mettre sur le marché cette molécule car elle la juge trop toxique. Quelques grammes suffisent, en effet, pour tuer un humain.
A partir de 1955, on utilise comme antipaludéen une molécule voisine de la chloroquine, l’hydroxychloroquine, qui sert aussi en rhumatologie. Ces deux molécules ne sont plus guère utilisées aujourd’hui comme antipaludéen sauf pour certains types particuliers. Comme chacun sait, des essais cliniques sont en cours pour évaluer leur action antivirale.
Il existe à ce jour d’autres aminoquinoléines, dont l’amodiaquine et la méfloquine (Lariam), qui sont actives pour traiter certains plasmodiums résistants aux autres molécules.
Il existe également plusieurs molécules actives contre la malaria, dont certaines ne sont pas des aminoquinoléines. Actuellement, une de ces molécules prometteuses est l’artémisinine. Extraite de l’absinthe chinoise, c’est une lactone comportant un groupe peroxyde. Malheureusement, cette molécule possède sept centres d’asymétrie, d’où un très grand nombre de stéréo-isomères, ce qui rend sa synthèse quasi impossible.
En raison de la résistance des plasmodiums aux nouvelles molécules qu’on leur oppose, la recherche d’antipaludéens, si l’on retient comme début les travaux de Pelletier et Caventou, dure depuis le début du XIXe siècle. Cette recherche de plus de deux cents ans n’aura probablement pas de fin, comme celle, encore plus complexe, concernant les virus.